Simulation Cell Relaxation
These notes describe the usage and functionality implemented in ONETEP for the calculation of the stress tensor and related quantities.
User guide
Keyword |
Type |
Default |
Description |
|---|---|---|---|
|
Task |
— |
Enable stress functionality. |
|
Logical |
|
Enable the calculation of
the stress tensor.
|
|
Logical |
|
Enable the calculation of
elastic constants
|
|
Logical |
|
Use the stress tensor to
optimise the cell parameters.
|
|
String |
|
Use assumed symmetry
to minimise calculations.
Values are
nosymm; 3D: cubicortho, tetra1, tetra2hexa3d, rhomb1, rhomb2;2D:
recta, squar1, squar2, hexa2dNOTE: Symmetry is assumed,
the code will not check if it’s correct!
|
|
Real |
|
Rescaling for cell volume.
Might be useful for 1D or 2D systems.
|
|
Logical |
|
Which rows/columns of the stress tensor
to compute. The flags match X Y Z.
|
|
Real |
|
Unitless strain parameter in finit difference
It controls how different the deformation
matrix is from the identity
|
|
Integer |
|
Maximum number of NGWF CG iterations for total
energy calculations of distorted cells needed
for the stress tensor.
|
|
Physical |
|
Convergence criterion for absolute change of
energy in cell relaxation.
|
|
Physical |
|
External pressure applied during cell.
relaxation
|
|
Physical |
|
Convergence criterion for absolute change of
pressure in cell relaxation
|
|
Real |
|
Convergence criterion for relative change of c
cell parameters in cell relaxation.
|
|
Integer |
|
Maximum number of iterations in cell relaxation |
|
Real |
|
Maximum step size for distortion in cell
relaxation.
|
|
Logical |
|
Atomic positions are relaxed together with cell
parameters.
|
The keywords table lists the input parameters that are available in
connection with the stress implementation. The main functionality can be
enabled by specifying the corresponding keyword (stress_tensor,
stress_elasticity or stress_relax) without providing additional
options. If the default values of the unspecified options are found to
be unsuitable for the target system they can be overridden with the
corresponding keyword in the input file.
The STRESS task gives access to all the features of the stress
implementation. It first performs a standard self-consistent calculation
for a reference unit cell (as given in the input file), and then
performs subsequent calculations according to the requested
stress-related quantities. This can be sped up quite considerably by
writing out the density kernel (write_denskern T) or Hamiltonian
(write_hamiltonian T) and NGWFs (write_tightbox_ngwfs T) files at
the end of the self-consistent calculation for the reference unit cell.
These should then be read for the calculations of the distorted
structures used in constructing the stress tensor (read_denskern T,
read_tightbox_ngwfs T and read_hamiltonian T). The code makes
sure that the distorted cell calculations do not overwrite these files;
if one is performing a cell relaxation calculation then these files can
be updated by the self-consistent calculation for each new trial unit
cell. Empirical observation: performing cell relaxation using only
write_tightbox_ngwfs T shows the most stable and robust behaviour
for the test calculations that I performed so far.
Calculation of the stress tensor
The definition for the stress tensor is
Here \(V\) is the volume of the unit cell, \(E\) is the total energy (of the unit cell), \(\sigma\) and \(\epsilon\) are the stress and strain tensors, respectively, and \(\alpha,\beta = x,y,z\). To convert to experimental units: energy divided by volume has units of pressure. The strain tensor describes a deformation of space away from a reference configuration: \(r_\alpha' = \left(\delta_{\alpha\beta} + \epsilon_{\alpha\beta}\right) r_\beta\) (Einstein summation convention) or \({\mathbf{r}}' = \left(1 + \epsilon\right) {\mathbf{r}}\). For more details see Section Background — Definition of stress for a crystal.
The stress tensor is calculated starting from a self-consistent
calculation for a reference unit cell, applying distortions to those
reference cell vectors, and using the corresponding total energy
differences to approximate the derivatives of the total energy with
respect to the distortion parameters. This is available through the
STRESS task by setting stress_tensor to T.
Here are two examples of distortion matrices:
These act on the matrix of cell vectors as e.g.
The parameter \(h\) is controlled by the keyword
stress_deformation_step. As \(|h| \ll 1\), if the calculation
for the distorted structure is started from the converged ground state
obtained for the reference structure it should converge in a handful of
iterations; stress_maxit_ngwf_cg can be used to control this and
avoid spending too many iterations on a calculation that may have
incorrect input and so takes too long to converge.
The stress tensor is a \(3\times3\) symmetric matrix, so it has 6
independent components. Finding all these components then requires 12
calculations for distorted cells (\(+h\) and \(-h\)), to form
the centred differences that approximate the total energy derivatives.
This can be alleviated if the unit cell is expected to have a definite
symmetry. For example, for a cubic crystal
\(\sigma_{xx} = \sigma_{yy} = \sigma_{zz} \neq 0\) and
\(\sigma_{xy} = \sigma_{yz} = \sigma_{zx} = 0\), so only two
distortions are needed (\(\epsilon_{xx}^+\) and
\(\epsilon_{xx}^-\)). The keyword stress_assumed_symmetry
controls the available options; see Table for a quick
conversion and Table I of Ref. for the notation and further details.
NOTE: Symmetry is assumed, the code will not check if it’s correct!
The stress tensor components only make sense if the corresponding
spatial directions have periodic boundary conditions, which is the case
of a bulk crystal. The keyword stress_components gives control over
this, and if any of its flags is F it will override
stress_assumed_symmetry to use nosymm. For example, for a 2D
material with periodic boundary conditions in \(x\) and \(y\)
and open boundary conditions (or a thick vacuum region) in the \(z\)
direction, respectively, one would specify T T F to skip calculation
(and usage) of information that involves the \(z\) direction. As
another example, consider a nanotube which is periodic in the \(z\)
direction; this would correspond to the flags F F T. Lastly, the
stress tensor should not be computed for something which is surrounded
entirely by vacuum, such as a molecule.
|
|
|
|
|
|
|
\(m\bar{3}\), \(m\bar{3}m\) |
\(mmm\) |
\(4/mmm\) |
\(4/m\) |
\(6/mmm\), \(6/m\) |
\(\bar{3}m\) |
\(\bar{3}\) |
|
|
|
|
|||
\(2mm\) |
\(4mm\) |
\(4\) |
\(6\), \(6mm\) |
Calculation of the elastic properties
This part is still being developed, it should not be used yet.
Optimisation of the cell parameters
The stress tensor can be used to optimise the cell parameters by
minimising the total energy with the STRESS task. This feature is
enabled by stress_relax T. It could also be integrated with the
GEOMETRYOPTIMIZATION task (not implemented so far).
Let us consider for simplicity that the atomic positions are fixed (relative to the unit cell vectors). We start from a unit cell specified by the vectors \(\{{\mathbf{a}}_1, {\mathbf{a}}_2, {\mathbf{a}}_3\}\) and want to calculate the optimised vectors \(\{{\mathbf{a}}_1^*, {\mathbf{a}}_2^*, {\mathbf{a}}_3^*\}\) that minimise the energy of the system, and so for which the stress tensor vanishes, \(\sigma_{\alpha\beta}(\{{\mathbf{a}}_i^*\}) = 0\). We can relate the known input cell parameters to the unknown optimised ones by a strain matrix,
We then Taylor expand the total energy in the strain parameters,
For the stress to vanish we evaluate the first derivative w.r.t. the strain parameters and set it to zero:
The first derivative of the total energy is approximated by centred differences, but this also provides enough information to approximate the diagonal part of the matrix of second derivatives (\(\alpha'\beta' = \alpha\beta\)). This leads to an approximate Newton-like step to solve for the strain parameters \(\epsilon_{\alpha\beta}\):
which is implemented as the cell relaxation method for fixed atomic positions.
In practice the optimisation of the cell parameters will not converge in
one iteration. The maximum number of iterations or trial unit cells is
controlled by stress_relax_max_iter. The magnitude of the
deformation is controlled by stress_relax_max_step, which ensures
that \(|\epsilon_{\alpha\beta}|\) does not exceed the specified
value. There are three convergence criteria that must be satisfied
simultaneously. stress_relax_energy_tol ensures that the change in
total energy per atom between consecutive trial unit cells is below a
given value, stress_relax_pressure_tol checks that the pressure of
the current unit cell is below a target value, and
stress_relax_acell_tol monitors the relative change in cell
parameters,
\(\frac{\max |\Delta a_{i\alpha}|}{\max |a_{i\alpha}|}\).
The atomic positions can also be optimised in tandem with the cell
parameters. This is enabled by the flag stress_relax_atoms. The
calculation will start by first optimising the atomic positions with
fixed cell parameters, and then it will create a guess at the cell
parameters to try next. If all the convergence thresholds for the cell
optimisation are met the calculation ends, otherwise it keeps iterating
by reoptimising the atomic positions and then making a new guess for the
cell parameters. The optimisation of the atomic positions proceeds in
the same way as with the GEOMETRYOPTIMIZATION task, and the
corresponding keywords/flags can be used to control the same aspects of
that process.
External pressure can included during cell relaxation using
stress_relax_pressure. This will be added to the diagonal elements
of the stress tensor which are not set to zero by the assumed symmetry
in the calculation. Positive values of pressure will lead to a
compression of the unit cell volume.
Background — Definition of stress for a crystal
These notes follow the original papers by Nielsen and Martin [Nielsen1983] [Nielsen1985] [Nielsen1985b] and the discussion in Chapter 3 of the book of Martin [Martin2008]. The definition for the stress tensor is
Here \(V\) is the volume of the unit cell, \(E\) is the total energy (of the unit cell), \(\sigma\) and \(\epsilon\) are the stress and strain tensors, respectively, and \(\alpha,\beta = x,y,z\). To convert to experimental units: energy divided by volume has units of pressure. \(1 \text{GPa} = 10 \text{kbar} = 10^9 j/m^3\), so it’s enough to convert the energy to Joule and the volume to cubic meters.
The strain tensor describes a deformation of space away from a reference configuration: \(r_\alpha' = \left(\delta_{\alpha\beta} + \epsilon_{\alpha\beta}\right) r_\beta\) (Einstein summation convention) or \({\mathbf{r}}' = \left(1 + \epsilon\right) {\mathbf{r}}\). The way this works is easiest to understand in one dimension and for a one-particle wave function \(\Psi(x)\), defined in the interval \([0,L]\) which is the unit cell for this example. Suppose that the unit cell is stretched to \([0,L']\) with \(L' = \left(1 + \epsilon\right) L\), and so the wave function will also be stretched to \(\widetilde{\Psi}(x')\) with the coordinate in the stretched unit cell being related to the starting one by \(x' = \left(1 + \epsilon\right) x\). This is illustrated in Fig. 10. The wave function at the stretched coordinate is almost the same as the wave function at the unstretched coordinate,
where \(C\) is a constant to be determined. [This is usually horribly confusing; a function returns a specified output for a given input, so if we want to know what is the value of the wave function \(\widetilde{\Psi}\) at \(x'\) we need to first transform that to the input for the wave function \(\Psi\) that will generate the correct output.] The remaining difference is that the wave function has to be normalised to the unit cell,
and so the complete relation is \(\widetilde{\Psi}(x') = \left(1 + \epsilon\right)^{-1/2} \Psi((1 + \epsilon)^{-1}x')\). Intuitively, the volume element is changed as \({\mathrm{d}}x' = \left(1 + \epsilon\right) {\mathrm{d}}x\), so the normalisation is adjusted to compensate.
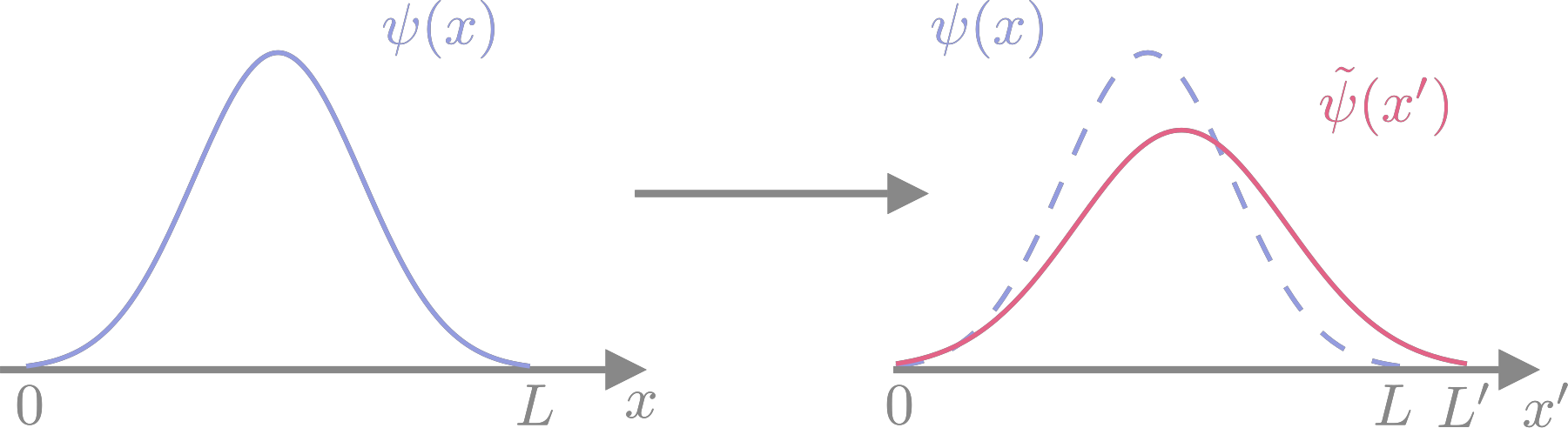
Fig. 10 One-dimensional example of coordinate stretching. Comparing the wave function in the stretched coordinate system to the one in the original coordinates shows that its centre (nuclear position) and its amplitude (normalisation) are both changed by the scaling
Moving now to the usual three-dimensional problem and to a crystalline setting, we first express the position vector in terms of the vectors defining the reference unit cell:
In Cartesian coordinates this looks like
The transformation defining the action of the strain tensor \(\epsilon\) on the crystal can then be converted into a deformation of the unit cell:
The strain tensor is assumed symmetric; a skew-symmetric part would represent a homogeneous rotation of the whole crystal which should leave the energy invariant. The elements of the strain tensor have a simple interpretation when the unit cell vectors are proportional to the unit vectors defining the cartesian axes (e.g., orthorhombic cell): the diagonal elements \(\epsilon_{\alpha\alpha}\) (\(\alpha = x, y, z\)) represent a stretching or compression along the respective unit cell vector or cartesian axis (angles between the unit cell vectors are preserved), while the off-diagonal elements represent shear (the angle between the involved pair of unit cell vectors changes).
It is also common to map the subscripts to integers (Voigt notation),
To understand how that factor of 2 propagates see the chapter on elasticity of Kittel’s book [Kittel2004] .
The strain can also be represented in a form which uses directly the unit cell vectors. Defining
we can write
This is in general not a symmetric tensor in Cartesian components, but we enforce \(\epsilon_{ij} = \epsilon_{ji}\). It also only makes life easier if it acts on the matrix of cell vectors from the left,
The different elements can then be interpreted as effecting a stretch/compression of the respective unit cell vector (\(\epsilon_{ii}\)) or shears (\(\epsilon_{ij}\) with \(i \neq j\)), which bring cell vectors \(i\) and \(j\) towards/away from each other.
To be investigated:
Computing the stress tensor
Ref. [Knuth2015] has nice derivations and a discussion of finite-difference tests in Section 4. For Projector Augmented Wave (PAW) specifics I looked at Ref. [Kresse1999] , but sadly they wrote “[…] it is also easy to evaluate the stress tensor. We will neither give the full derivation nor the final results here, as the expressions are rather cumbersome and difficult to write in a compact form.”
The straightforward numerical calculation by finite differences proceeds in the obvious way (here using the central difference formula):
where the total energies are obtained from self-consistent calculations for the deformed unit cell with only one finite element in the strain tensor. The atomic positions should either be given in internal coordinates or otherwise their Cartesian coordinates have to be scaled. The unitless \(h\) has to be tuned via numerical tests, but values \(h < 0.01\) are mentioned as reasonable in Ref. [Knuth2015] ; for example Nielsen and Martin used \(h = 0.004\) [Nielsen1985b]. To fully populate the stress tensor using the central difference scheme for the general case one then requires 12 self-consistent calculations.
References
Nielsen and R. M. Martin, First-principles calculation of stress, Phys. Rev. Lett. 50, 697 (1983).
Nielsen and R. M. Martin, Quantum-mechanical theory of stress and force, Phys. Rev.B 32, 3780 (1985).
Nielsen and R. M. Martin, Stresses in semiconductors: Ab initio calculations on Si, Ge, and GaAs, Phys. Rev. B 32, 3792 (1985)
Martin, Electronic structure, 1st ed. (Cambridge Univ. Press, Cambridge, 2008)
Kittel, Introduction to Solid State Physics, 8th ed. (Wiley, 2004).
Knuth, C. Carbogno, V. Atalla, V. Blum, and M. Scheffler, All-electron formalism for total energy strain derivatives and stress tensor components for numeric atom-centered orbitals, Comput. Phys. Commun. 190, 33 (2015)
Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B 59, 1758 (1999).